
在过去的几年里,SARS-CoV-2在全球范围内传播,导致了一场全球大流行。迄今为止,这一大流行已感染5.9亿多人,造成640万多人死亡,更严重的是,恢复期患者出现了各种持续和延长的后遗症,带来了更复杂的医疗问题。尽管有疫苗可用,但不可避免地会出现难以治愈的严重病例。因此,根治仍然需要治疗性抗体。
然而,抗体设计是一项复杂的蛋白质设计任务,因为SARS-CoV-2家族以高度变异性而闻名。目前已知的变种有10多种,其中阿尔法、贝塔、伽马、德尔塔突变导致了全球传播,奥密克戎现在正在肆虐。快速的突变给传统的理性设计带来了新的挑战,即需要考虑多个氨基酸对结合位点的影响。许多抗体设计在应用于SARS-CoV-2家族的新变种时,已被证明敏感性降低,甚至失去了中和活性,这给该领域留下了一个棘手的问题。
蛋白质-蛋白质相互作用是基于抗体的治疗的基本机制,因此,增强抗体的特异性亲和力成为新药开发其中一个最重要的关键点。通过传统的基于湿实验室的策略,科学家引入随机突变并进一步筛选具有更好功能特性的抗体,然而,这样的实验方法非常耗时,更重要的是无法覆盖氨基酸的大组合空间。为了解决这个问题,华深智药推出了一种有用的计算抗体优化工具,并将其应用于优化针对多种严重急性呼吸综合征冠状病毒2(SARS-CoV-2)变体的中性抗体。

我们训练了一个几何深度学习模型,可以有效地增强抗体亲和力,以实现更广泛和更有效的中和活性。基于注意力的网络能够捕获模型中存在的交互特征。在我们的模型中,该特性被完美地利用来提取抗原和抗体之间的残基间的相互作用特征。
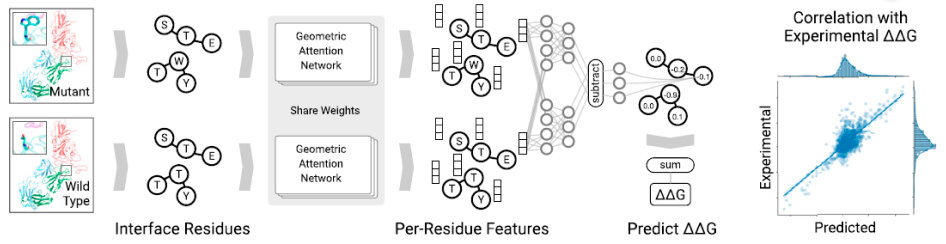
基于这种几何嵌入,网络需学习识别蛋白质界面附近有助于结合亲和力的关键残基对,从而使我们能够测量突变的影响 ,即自由能变化(ΔΔG)。为了寻找有利的互补决定区域(CDR),我们用CDR模拟了预测复杂结构的计算机集合。 自由能变化(ΔΔG)被设置为深度神经网络中的目标函数,有助于搜索最佳候选者。
我们证明了我们的模型在人中和抗体P36-5D2上的实用性,该抗体对SARS-CoV-2 Alpha、Beta和γ具有很强的效力,但对Delta变体没有效力。我们的模型在优化这种中和抗体方面表现出色。在实验验证中,预测的最佳CDR序列确实有助于提高优化的抗体对Delta变体的结合亲和力,同时保持对Alpha,Beta和Gamma的活性。通过建模和实验验证的迭代过程,我们能够获得六种优化的抗体,对多种变体的效力显著提高约10至600倍,也对奥密克戎提供了初步有希望的研究。
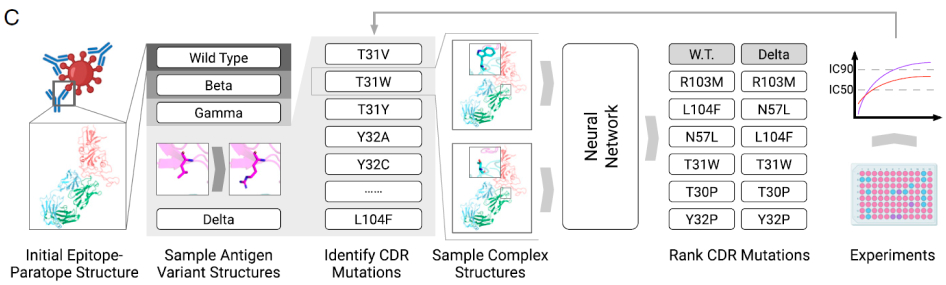
这种用于抗体优化的AI解决方案在各个方面都扩展了传统实验方法的局限性。与随机诱变相比,CDR的计算模拟在很大程度上扩大了候选库。更重要的是,实验突变有时会带走抗体的关键功能残基,同时带来潜在的改进。计算策略可以避免这个问题,从而寻找最佳候选者。
我们模型的出色性能和突出特性体现了深度学习神经网络在抗体优化方面的能力及其在工程其他蛋白质分子中的潜在应用。这种计算方法不仅能带来更多可以开发成有前途的抗体药物的候选蛋白质,而且还能打开人工蛋白质设计的视野。
“这项新工作标志着人工智能的一个里程碑:通过使用新颖的计算机驱动方法来改进传统的生物产品,扩展传染病治疗中的传统湿实验室方法。”
ΔΔG预测的代码已经在Github上发布:https://github.com/HeliXonProtein/binding-ddg-predictor